نورونهای موجود در قاعده مغز (Base of skull) و شاخ جلویی نخاع (Ventral Root) از نوع موتورنورونها بوده در ایجاد حرکات بدن نقش اصلی را دارند. از بین رفتن تدریجی این سلولها بعلت عدم تعادل در آپوپتوزسلولهای شاخ قدامی نخاع، سبب ایجاد بیماری آتروفی عضلانی – نخاعی (Spinal muscular atrophy (SMA میشود.
SMA واژهای برای توصیف، گروهی هتروژن (از نظر علائم بالینی و علت بروز) میباشد. این بیماری از اختلالات ضعف عضلانی پیشرونده است و بیشتر یک بیماری وراثتی قلمداد میشود. بدین معنی که در 95 درصد موارد، مبتلایان به SMA دچار اختلال در ژنی بنام SMN1 هستند. این ژن پروتئینی را کد میکند موسوم به پروتئین بقا. در صورت عدم حضور نورونهای حرکتی، که نقش مهمی در عملکرد صحیح نورونها به عهده دارند، سلولهای عصبی از بین میروند. ژنی دیگر به نامSMN2 نیز پروتئین SMN تولید میکند ولی درواقع تنها حدود 10 تا 15٪ از پروتئین ساخته شده از این ژن عملکردی است. افراد می توانند به طور معمول، بین صفر تا هشت نسخه از ژن SMN2 داشته باشند و هرچه تعداد کپی از این ژن در شخص بیشتر باشد، بخاطر بیشتر بودن عملکرد پروتئین SMN، روند بیماری خفیفتر خواهد بود. برای مثال داشتن سه یا چند نسخه از ژن SMN2 با تظاهرات بالینی خفیف بیماری همراه است. از نظر کروموزومی، هر دو این ژنها بر روی کروموزم 5 واقع شدهاند و به صورت اتوزوم مغلوب به ارث میرسند. بدیهی است، نرخ بروز SMA در فرزندان دختر و پسر یکسان خواهد بود.
تشخیص:
کم هزینهترین راه برای تشخیص بیماری SMA انجـام الکترومایوگرافی یا نوار عصب و عضله برای بیمار است. به دلیل اینکه این بیماری عصبی-عضلانی میباشد تهیه نوار عصب و عضله میتواند نمای خاصی از این بیماری را نشان دهد. تشخیص قطعی SMA بر اساس روشهای ژنتیک مولکولی صورت میگیرد. در حدود ۹۵ تا ۹۸٪ افراد مبتلا، دچار حذف هموزیگوت دراگزون ۷ و ۸ ژن SMN1 هستند. به همین دلیل، در روش مستقیم برای تشخیص این بیماری، بیماران از نظر حذف این اگزونها در ژن SMN1 مورد بررسی قرار میگیرند. روش غیر مستقیمی نیز وجود دارد که در آن از STRها به طور همزمان جهت تشخیص بیماری استفاده میشود. با بهرهگیری از همین دو روش، میتوان به تشخیص پیش از تولد نوزادان نیز دست یافت.
پـاتوژنز:
وضعیت ژن SMN بر فرم بیماری مؤثر است. به بیان دیگر، شدت و زمان بروز علائم اولیه بیماری SMA، وابسته به میزان پروتئین طبیعی این ژن است که در سلولها وجود دارد. با افزایش سطح میزان پروتئین طبیعی، شدت علائم کمتر و سن اولیه بروز تظاهرات اولیه بیماری، بالاتر خواهد بود.
آتروفی عضلانی – نخاعی بر اساس میزان شدت و زمان بروز علائم اولیه بیماری، به چهار نوع شدید (SMAI), متوسط (SMAII) و خفیف (SMAIII/IV) دستهبندی میشود.
١– SMA (Werdnig Hoffmann syndrome)
شایعترین نوع بیماری، نوع شدید یا ١ SMA است. به آن سندرم بچه شُل یا SMA نوع صفر هم میگویند. سن رایج آغاز بیماری، از بدو تولد تا 6 ماهگی است. تظاهرات بالینی این فرم شامل ضعف عضلانی, فلج ناحیه حلق، فرورفتگی قفسه سینه، نارسائی تنفسی است که از ابتدای تولد شروع شده و معمولاً در سال اول زندگی منجر به مرگ می شود.
https://care.togetherinsma.eu/en/home/sma-in-infants-and-children/sma-signs-and-symptoms.html

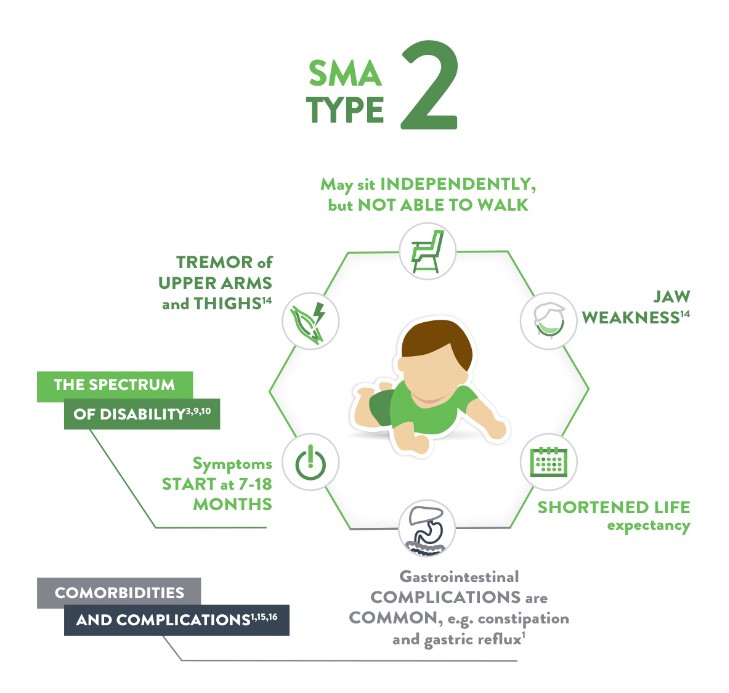
SMA -۲ آتروفی عضلانی نخاعی تیپ ٢ (Dubowitz syndrome)
از نظر شدت، از١SMA خفیفتر است. علائم بین ماههای 6 تا 18 ماه پس از تولد بروز مییابند. همانند تیپ ١ ضعف عضلانی و هیپوتونی ویژگی های اصلی بیماری هستند. این کودکان اگرچه بدون کمک قادر به نشستن هستند، اما هرگز قادر نیستند به شکل مستقل و بدون حمایت جابجا شوند. پیشرفت آهسته بوده و بیشتر کودکان تا اوایل دوران بلوغ زنده میمانند.
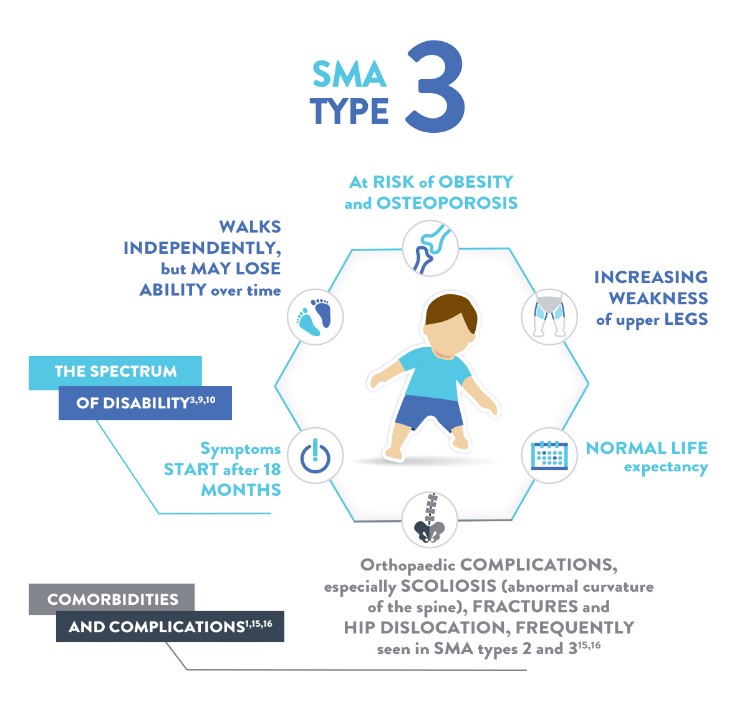
٣- SMA آتروفی عضلانی نخاعی تیپ ٣ (Kugelberg–Welander SMA)
این نوع شکل نسبتاً خفیف از این بیماری در دوران کودکی یا نوجوانی است و سن بروز آن پس از 18 ماهگی بوده و همه بیماران قادرند بدون حمایت راه بروند. پیشرفت آهسته ضعف عضلانی منجر به این میشود که بسیاری از افراد مبتلا در اوایل دوران بلوغ مجبور به استفاده از صندلی چرخ دار گردند. بقای طولانی مدت بیماران این تیپ با عفونت مکرر تنفسی و بروز اسکولیوز (خمیده شدن طرفی ستون مهره ها) که بوسیلهی ضعف عضلات نخاعی ایجاد میشود، به خطر میافتد.
٤- SMA آتروفی عضلانی نخاعی تیپ ٤ (بالغین)
به آن٣ SMA تأخیری یا دیر هنگام هم میگویند. معمولاً پس از 30 سالگی و بهصورت خستگی، ضعف تدریجی عضلات و اشکال در تنفس بروز مییابد. بیشتر عضلات پروگزیمال اندام (بازوها و رانها) را درگیر میکند و منجر میشود که فرد برای تحرک، وابسته به صندلی چرخدار شود. عوارض دیگر نادر است و امید به زندگی تغییری نمیکند.
مقایسه کارایی دو تکنیک Whole-exome Sequencing و RNA-Seq در تشخیصی بیماریهای عصبی-عضلانی
استفاده از توالی یابی کل اگزوم[1] برای تشخیص بیماری های نادر معمولاً 40 تا 50 درصد نتیجه میدهد. ناهمگنی مکان[2] یا ناهمگنی فنوتیپی[3] از جمله موانعی است که میزان موفقیت در تشخیص دقیق بیماران مبتلا به اختلالات عصبی عضلانی (NMDs) را تحت تأثیر قرار میدهد. در یکی ازمقالاتی که اخیراً به عنوان پرخوانندهترین مقاله درنشـریه J Med Genet انتخاب شدهاست، نگارنده به بررسی سودمندی توالی یابی رونوشت[4] به عنوان یک رویکرد مستقل در تشخیص NMD پرداخته است.
محققان کرهای در این مطالعه، با هدف بررسی توانایی این تکنیک برای تشخیص انواع واریانتهای بیماریزا[5]، مولکولهای RNA استخراج شده از بافتهای عضلانی 117 بیمار مشکوک به NMD مندلی[6] را با استفاده از روش RNA-Seq مورد ارزیابی قرار دادند. پیرایش نابجا [7]RNA و تنوع تعداد کپی ژنومی (CNVs)، برای شناسایی عوامل ژنتیکی جانبی در ایجاد NMD مورد بررسی قرار گرفتند. برای بررسی وضعیت پیرایش نابجا در mRNA ژن دیستروفین[8] (DMD)، از آنتی سنس الیگونوکلئوتید (ASOs)[9] استفاده شد. محققان در این مطالعه، منحصراً با استفاده از آنالیز ترانسکریپتوم سلولهای عضلانی، موفق به شناسایی (38.1٪) واریانتهای پاتولوژیک شدند که در مقایسه با آنالیز اگزوم (34.9٪)، نرخ تشخیصی بالاتری را از خود نشان میدهد. کشف واریانتهای ژنتیکی که سبب پیرایش نابجا در mRNA میشوند، این امکان را فراهم میآورد تا با استفاده از روش ASOs روی سلولهایی که از بیمار گرفته شده، یک رویکرد درمانی متناسب با هر بیمار را در پیش بگیریم. از دیگر مزایای استفاده از دادههای حاصل از RNA-Seq بر exome sequencing این است که این روش این امکان را فراهم میآورد تا با استفاده از پروفایل های بیان ژن متمایز که با پارامترهای بالینی مطابقت دارد، خوشه بندی نمونه را داشته باشیم. و در پایان، نگارنده این مقاله اینگونه استنباط کرده که RNA-Seq دارای نرخ تشخیص بالاتری در مورد بیماران مشکوک به اختلالات عصبی عضلانی مندلی است و اطلاعاتی از وضعیت بیماری زایی ارائه میدهد که به راحتی از طریق تجزیه و تحلیل اگزوم قابل دسترسی نیست.
Hong, S.E., et al., Transcriptome-based variant calling and aberrant mRNA discovery enhance diagnostic efficiency for neuromuscular diseases. J Med Genet, 2022. 59(11): p. 1075-1081.
درمان SMA با استفاده از داروی Zolgensma
پیش نویس نهایی دستورالعملNICE [10] ژن درمانی را برای درمان آتروفی عضلانی نخاعی (SMA) تایید می کند.
آتروفی عضلانی نخاعی (SMA) یک بیماری عصبی عضلانی پیشرونده نادر است که در اثر یک جهش ژنتیکی ایجاد میشود. این بیماری با از دست دادن تدریجی نورونهای حرکتی که باعث ضعف عضلانی، از دست دادن تدریجی حرکت و مشکل در تنفس و بلع می شود همراه است. درمان جدید و بالقوه موثر نوزادان مبتلا به SMA با استفاده از تکنیک ژن درمانی یکباره[11] قرار است به گرانترین درمان مورد تایید NICE تبدیل شود.
در پیش نویس نهایی راهنمای NICE که 4 ژوئن 2021 منتشر شد، درمان 1.79 میلیون پوندی Zolgensma (که همچنین به نام onasemnogene abeparvovec نیز شناخته میشود و توسط شرکت دارویی Novartis Gene Therapies ساخته شده) برای نوزادان تا 12 ماه مبتلا به نوع ١ SMA را توصیه شده است.
١ SMA یکی از شدیدترین اشکال آتروفی عضلانی نخاعی بوده و امید به زندگی افراد مبتلا به آن معمولا کمتر از 2 سال است. سالانه حدود 65 نوزاد با SMA در انگلستان متولد میشوند که حدود 60 درصد از آنها با نوع ١ SMA تشخیص داده می شوند.
علیرغم قیمت بسیار بالای این دارو، همچنان در مورد “مزایای بلندمدت” Zolgensma تردیدهایی وجود دارد، موضوعی که در مورد بسیاری از درمانهای جدید برای بیماریهای بسیار نادر ممکن است مصداق داشته باشد.
[1] Whole-exome Sequencing
[2] locus heterogeneity
[3] phenotypic heterogeneity
[4] transcriptome sequencing
[5] pathogenic variants
[6] Mendelian NMD
[7] Aberrant splicing
[8] Dystrophin
[9] Antisense oligonucleotides (ASOs).
[10] National Institute for Health and Care Excellence
[11] one-off gene therapy


Leave feedback about this