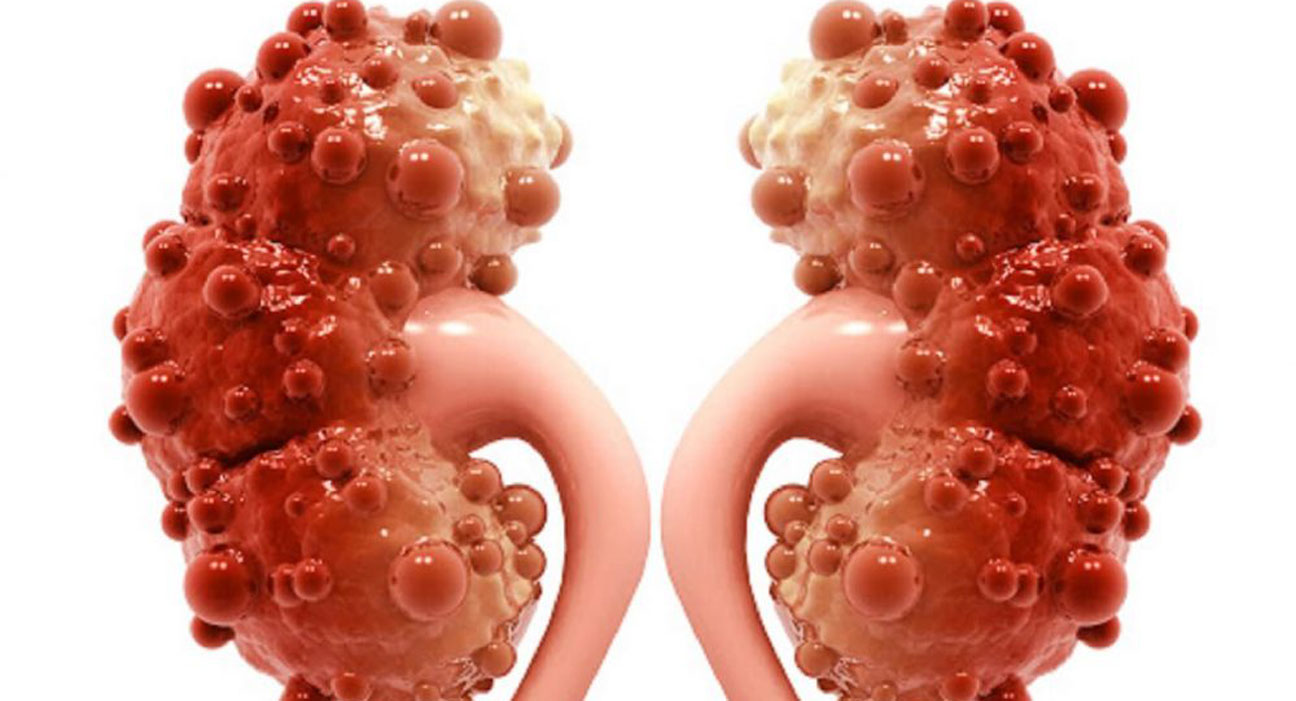
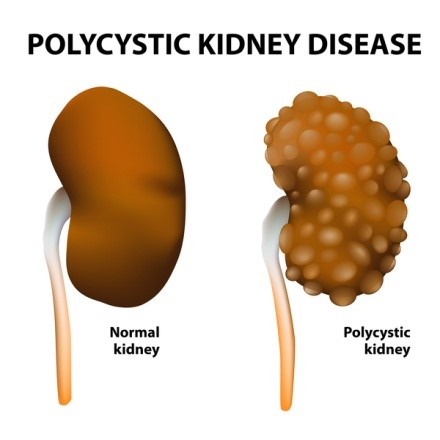
بیماری کلیهی پلیکیستیک عموما باعث اختلال در عملکرد کلیهها میگردد. در این بیماری کیسههایی پر از مایع که کیست نامیده میشوند در کلیه تشکیل میشود. وجود این کیستها عملکرد کلیه در تصفیه خون از مواد زائد را مختل میکند. رشد کیستها باعث بزرگتر شدن کلیهها میگردد. این کیستها علاوه بر کلیهها ممکن است در سایر اندامها بویژه کبد نیز وجود داشته باشند.
مهمترین علائم بیماری کلیهی پلیکیستیک شامل: فشار خون بالا، درد در پشت یا پهلوها، وجود خون در ادرار، عفونتهای مکرر مجاری ادراری، سنگ کلیه و ناهنجاریهای قلبی میباشد. علاوه بر این افراد مبتلا به بیماری کلیهی پلیکیستیک در معرض ابتلا به آنوریسم آئورت و یا آنوریسم عروق مغزی میباشند.( آنوریسم نوعی برآمدگی غیر طبیعی در رگهای خونی است که در صورت پاره شدن تهدید کننده حیات فرد میباشد.)
بیماری کلیهی پلیکیستیک به دو شکل اصلی دیده میشود و تفاوت آنها در سن شروع بیماری و نحوه توارث آنها میباشد. در فرم اتوزومال غالب که ADPKD نیز نامیده دارد، نشانههای بیماری معمولاً در بزرگسالی دیده میشوند، هرچند ممکن است از بدو تولد یا کودکی کیستهایی در کلیه وجود داشته باشد. بسته به علت ژنتیکی بیماری شکل اتوزومال غالب بیماری به نوع یک و دو تقسیم بندی میشود. شکل اتوزومال مغلوب بیماری کلیهی پلیکیستیک بسیار نادرتر است و اغلب در اوایل زندگی کشنده است. علایم و نشانههای این نوع از بیماری معمولاً در بدو تولد یا در اوایل نوزادی رویت میشوند.
بیماری کلیهی پلیکیستیک یک اختلال ژنتیک نسبتاً شایع است. در ایالات متحده آمریکا یک نفر در هر 500 تا 1000 نفر به این بیماری مبتلا میباشد. نوع اتوزومال مغلوب شیوع کمتری دارد.
ژنتیک بیماری PKD
جهش در ژنهای PKD1 ، PKD2 و PKHD1 باعث بیماری کلیهی پلیکیستیک میشود. جهش در ژنهای PKD1 و PKD2 معمولاً نوع اتوزومال غالب این بیماری را ایجاد میکنند و به ترتیب باعث ایجاد ADPKD نوع 1 و نوع 2 میشوند. عملکرد محصول پروتئینی این ژنها، بطور کامل شناخته نشدهاند. ولی احتمالاً در انتقال سیگنال از خارج سلول به داخل هسته نقش دارند. برای عملکرد طبیعی کلیه عملکرد این دو پروتئین با هم حائز اهمیت میباشد. جهش در ژن PKD1 یا PKD2 باعث تشکیل هزاران کیست میشود که عملکرد کلیه را مختل میکند. افراد دارای جهش در ژن PKD2 ، به ویژه زنان، معمولاٌ نسبت به افراد دارای جهش در ژن PKD1 فرم خفیفتر بیماری را دارند. جهش در ژن PKHD1 باعث بیماری کلیهی پلیکیستیک از نوع اتوزومال مغلوب میشود. نقش احتمالی پروتئین حاصل از این ژن نیز انتقال سیگنالهای شیمیایی از خارج سلول به هسته میباشد. در صد بسیار کمی از موارد بیماری کلیهی پلیکیستیک منشا ژنتیکی ندارند و به این موارد کلیهی پلیکیستیک اکتسابی گفته میشود.
ژن PKD1
این ژن پروتئینی به نام پلی سیستین 1 را کد میکند. این پروتئین دارای یک ناحیه بزرگ ان ترمینال خارج سلولی، نواحی گذرنده از غشاء متعدد و یک ناحیه سی ترمینال سیتوپلاسمی است. پلی سیستین یک پروتئین اینتگرال است و در تنظیم غشایی کاتیون کلسیم و هموستاز کلسیم داخل سلولی نقش دارد. این پروتئین در نمو توبولار کلیه نقش دارد و جهش در این ژن باعث ایجاد کلیهی پلیکیستیک اتوزومال غالب نوع یک میشود. این ژن دارای شش سودوژن میباشد.
ژن PKD2
پروتئین حاصل از این ژن پلی سیستین 2 نام دارد و قبل از تولد در کلیهها و در بالغین در بافتهای مختلفی دیده میشود. این پروتئین احتمالاً بعنوان یک کانال در غشای سلولهای کلیه عمل میکند و انتقال کاتیونها بویژه یونهای کلسیم را به داخل بر عهده دارد.
ژن PKHD1
این ژن، پروتئینی به نام فیبروسیستین یا پلیداکتین را تولید میکند که در سلولهای کلیوی جنینی و بالغین دیده میشود و به میزان کمتری در سلولهای کبد و پانکراس نیز بیان میشود. این پروتئین نیز در انتقال سیگنالهای خارج سلولی به داخل سلول نقش دارد. علاوه بر این ممکن است در اتصال سلولها به یکدیگر و رشد و تقسیم سلولها نیز دخالت داشته باشد.
سایر ژنها: علاوه بر ژنهای فوق، در سالهای اخیر ژنهای دیگری نیز مورد توجه قرار گرفتهاند که جهشهای موجود در این ژنها نیز میتوانند در ایجاد نوع غالب یا مغلوب بیماری کلیهی پلیکیستیک نقش داشته باشند. تعدادی از این ژنها و نوع توارث آنها در جدول زیر ارائه شدهاند.
| Genes | Associated phenotypes | Inheritance |
| DNAJB11 | Autosomal dominant polycystic kidney disease | AD |
| DZIP1L | Polycystic kidney disease 5 | AR |
| GANAB | Polycystic kidney and/or polycystic liver disease 3 | AD |
| HNF1B | Renal cell carcinoma, nonpapillary chromophobe, Renal cysts and diabetes syndrome | AD |
| JAG1 | Alagille syndrome | AD |
| LRP5 | Van Buchem disease, Osteoporosis-pseudoglioma syndrome, Hyperostosis, endosteal, Osteosclerosis, Exudative vitreoretinopathy, Osteopetrosis late-onset form type 1, LRP5 primary osteoporosis | AD/AR/Digenic |
| NOTCH2 | Alagille syndrome, Hajdu-Cheney syndrome | AD |
| PKD1 | Polycystic kidney disease | AD |
| PKD2 | Polycystic kidney disease | AD |
| PKHD1 | Polycystic kidney disease | AR |
| PRKCSH | Polycystic liver disease | AD |
| SEC61A1 | Hyperuricemic nephropathy, familial juvenile 4 | AD |
| SEC63 | Polycystic liver disease | AD |
تستهای ژنتیکی تشخیص کلیه پلی کیستیک
بیماری کلیهی پلیکیستیک عمدتاً توسط دو ژن PKD1 و PKD2 ایجاد میشوند. در صورت وجود سابقه خانوادگی ADPKD ، بندرت از تستهای ژنتیکی استفاده میشود و معمولاً به تشخیصهای مبتنی بر تصویربرداری بسنده میکنند. علاوه بر این غربالگری جهشهای ژن PKD1 با توجه به بزرگی و پیچیدگی ژن و وجود چندین سودوژن همیشه با چالش مواجه بودهاست. در حال حاضر از تکنیکهای مبنتی بر PCR و توالییابی، Next Generation Sequencing(NGS) و در موارد کمتر MLPA برای تشخیص جهشهای مرتبط با بیماری کلیهی پلیکیستیک استفاده میشود. در صورت وجود سابقه خانوادگی از Linkage Analysis نیز میتوان برای تشخیص بیماری استفاده کرد.
اندیکاسیونهای تشخیص ژنتیکی بیماری کلیهی پلیکیستیک
- مشکوک به ADPKD بدون سابقه خانوادگی آشکار
- مشکوک به ADPKD با داشتن نتایج تصویر برداری دارای ابهام
- تایید یا رد ADPKD در افراد جوان در معرض خطر
- ارزیابی اهدای کلیه
- تشخیص ژنتیکی قبل از تولد یا قبل از انتقال جنین به رحم(PND & PGD)



Leave feedback about this